Breaking the Loop: The Cholesterol-Inflammation Fusion Hypothesis and What It Means for Your Heart

Breaking the Loop: The Cholesterol-Inflammation Fusion Hypothesis and What It Means for Your Heart
A CardioAdvocate Insight
The CardioConundrum: When "At Goal" Isn't Enough
Meet Linda. She's 62 years old and carries a little extra weight around the middle — not unusual for someone with her history. Eighteen months ago, she had an acute coronary syndrome event that changed her life and sent her cardiologist scrambling to rebuild her cardiovascular defenses. Since then, she's lost about 12 pounds through diet and exercise changes, which also helped bring her A1c down. On paper, the intervention worked beautifully. Her LDL cholesterol plummeted to 48 mg/dL — well below the 70 mg/dL target for secondary prevention. Her hemoglobin A1c sits at 6.2%, down from 7.1% at the time of her event, on metformin and empagliflozin (an SGLT2 inhibitor — standard of care for a diabetic with known cardiovascular disease). Her blood pressure runs 128/78 mmHg on a single beta-blocker. She takes her medications faithfully. She walks most days. Her husband says she's doing everything right.
And yet, last week, Linda walked into the clinic with new exertional chest pain. Her cardiologist ordered a nuclear stress test, which revealed a new area of ischemia — reduced blood flow to a region of the heart not involved in her original event. She was referred for a coronary angiogram, which confirmed progression of disease in a previously non-culprit vessel. A stent was placed. Linda had just experienced a recurrent coronary event.
This is the patient who falls through the cracks of single-pathway thinking. Linda represents a truth that decades of lipid-lowering trials have revealed but that we — as a field — haven't quite absorbed: LDL at goal does not mean risk eliminated. It means risk reduced. For many patients, something else continues to drive the disease forward. Something that may not always be visible on a standard lipid panel — or even on a standard inflammatory marker.
When Linda's cardiologist ordered a high-sensitivity C-reactive protein test, the result was 2.5 mg/L. Not dramatically elevated. Not the kind of number that sets off alarm bells. But here's what makes this interesting — and provocative: 2.5 mg/L falls in what most guidelines call "average" to "borderline high" risk. It's the kind of number that gets glossed over. And yet Linda just had a recurrent event despite doing everything right.
Her triglycerides were 210 mg/dL — a marker that often travels with the atherogenic triad of elevated triglycerides, low HDL, and increased small dense LDL particles. She was carrying some visceral adiposity — what we sometimes call adiposopathy, or "pissed off fat" — which likely contributed to some of her CRP elevation. But was adiposopathy the whole story? Or was something deeper going on — inflammation at the vessel wall level that CRP doesn't fully capture?
This is where the fusion hypothesis begins — and where Linda's story gets more nuanced than a single lab value can explain.
Flying Under the Radar: The False Sense of Security
The evidence has been staring us in the face. PROVE IT-TIMI 22 compared intensive LDL lowering (to 62 mg/dL) against standard therapy. The intensive group had better outcomes — but not as much better as the LDL reduction alone would predict. IMPROVE-IT, which added ezetimibe to statin therapy and pushed LDL even lower, showed that each additional 20 mg/dL reduction in LDL prevented about one additional event per 50 patients treated. That's real. But it's also a sobering reminder that cholesterol lowering — even aggressive cholesterol lowering — leaves much of the disease intact.
FOURIER, which enrolled over 27,000 patients on statin therapy and randomized them to PCSK9 inhibitor versus placebo, showed similar patterns. The PCSK9 inhibitor reduced LDL cholesterol by an additional 56% beyond statin therapy alone. Patients achieved LDL levels in the 30s and even 20s. And yes, they had fewer events. But the benefit seemed to plateau — and the question no one quite wanted to ask began to surface: Are we hitting a ceiling of what cholesterol-lowering alone can achieve?
This question intensified with CANTOS, the pivotal anti-inflammatory trial led by Paul Ridker. Here was direct proof that inflammation could be targeted independently of cholesterol. When researchers gave patients with prior heart attacks and elevated hs-CRP a specific inhibitor of IL-1β signaling, those patients had fewer recurrent events. The inflammation hypothesis — long suspected, now proven — could move from theory to practice.
But CANTOS came with a price. Patients treated with the anti-inflammatory agent had a higher risk of fatal infection and sepsis. The trial was halted prematurely. The message was clear: dampening one arm of the immune system has consequences. We can lower inflammation, but we cannot do it without cost.
For more on the inflammation story, see our deep dive: "The Itch That Rashes: Inflammation and Your Heart." And for a detailed look at EPA and fish oil mechanisms, read "Something Smells Fishy: EPA, DHA, and Fish Oil."
The Fusion Hypothesis: Two Fires, One Building
The cholesterol-inflammation fusion hypothesis, recently articulated by researchers like Zeng and colleagues in JACC Asia, proposes something deceptively simple: cholesterol and inflammation are not two separate problems. They are two arms of a single, self-reinforcing cycle. Breaking one arm slows the disease. Breaking both may stop it.
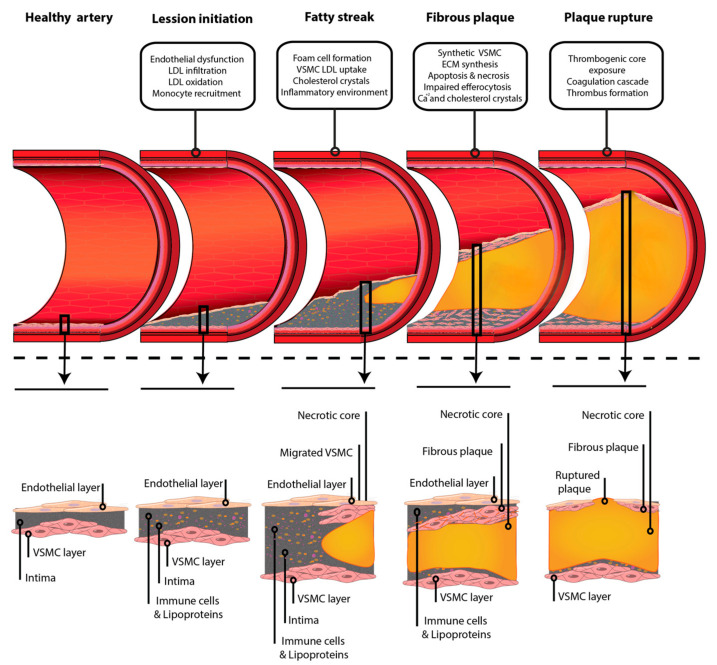
Figure 1: Atherosclerotic plaque progression — from healthy artery to plaque rupture, highlighting the interplay of lipid accumulation, foam cell formation, and inflammatory remodeling at each stage. Source: Jebari-Benslaiman S, et al. "Pathophysiology of Atherosclerosis." Int J Mol Sci. 2022;23(6):3346. PMC8954705. Used under CC BY 4.0 license.
Arm 1: Cholesterol Drives Inflammation
This arm is well-established. When LDL cholesterol accumulates in arterial walls, it undergoes oxidation. Oxidized LDL (ox-LDL) is recognized by scavenger receptors on macrophages, which eagerly take it up and transform into foam cells — the architectural foundations of atherosclerotic plaque. But the process doesn't stop there.
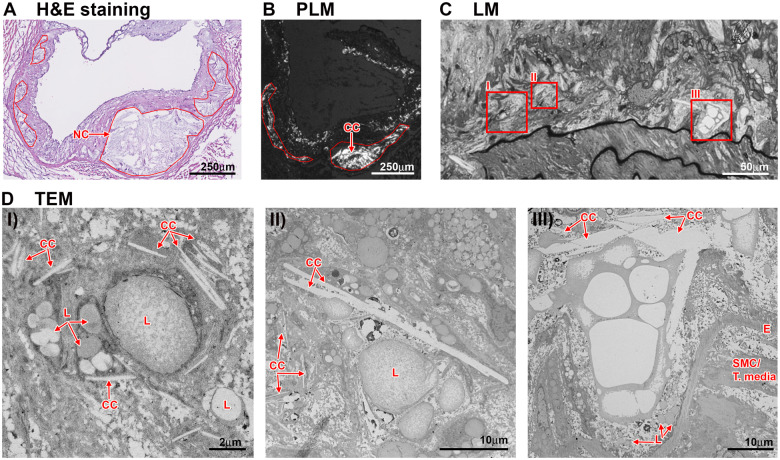
Figure 2: Cholesterol crystals within the necrotic core of atherosclerotic plaque — visualized by polarized light microscopy and transmission electron microscopy. These needle-shaped and plate-shaped crystals physically rupture cell membranes and activate the NLRP3 inflammasome, the molecular trigger at the heart of the fusion hypothesis. Source: Baumer Y, et al. "Cholesterol crystals and atherosclerosis." Eur Heart J. 2020;41(24):2236-2239. PMC7850045. Used under CC BY 4.0 license.
Inside the foam cell, cholesterol can crystallize. These cholesterol crystals physically activate the NLRP3 inflammasome, a molecular pattern-recognition complex that responds to danger signals. When NLRP3 activates, it releases large amounts of IL-1β, a potent pro-inflammatory cytokine. Additionally, oxidized and aggregated forms of LDL can signal through TLR4 (toll-like receptor 4), triggering additional inflammatory cascades that amplify the immune response.
This is cholesterol sparking inflammation. It's the first fire.
Arm 2: Inflammation Traps Cholesterol
But here's where it gets interesting — and dangerous. Once inflammation is established, it doesn't just sit passively. It actively suppresses the body's ability to clear cholesterol.
High levels of IL-1β, TNF-α, and IL-6 directly suppress the expression of ABCA1 and ABCG1 transporters — the molecular pumps that allow macrophages to eject cholesterol and return it to the bloodstream for HDL-mediated reverse cholesterol transport. When these transporters are suppressed, cholesterol becomes trapped inside the macrophage. The inflammatory environment also creates dysfunctional HDL particles — lipoproteins that have lost their ability to accept cholesterol from peripheral tissues and transport it back to the liver.
Inflammation also damages the endothelium, the delicate layer of cells lining the arterial wall. A damaged endothelium is more permeable — more LDL can infiltrate and accumulate. And as inflammation drives the production of matrix metalloproteinases, the fibrous cap that normally seals off plaque becomes thin and fragile, vulnerable to rupture.
This is inflammation trapping cholesterol. It's the second fire — and together, the two fires feed each other.
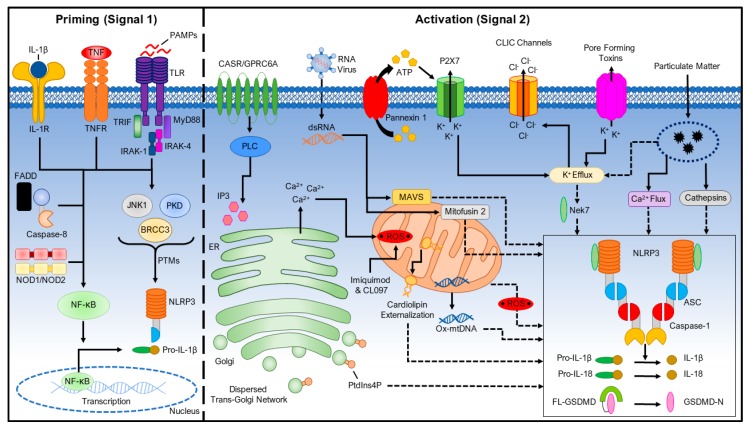
Figure 3: The two-signal model for NLRP3 inflammasome activation. Signal 1 (priming) activates NF-κB and upregulates NLRP3 and pro-IL-1β. Signal 2 (activation) — triggered by cholesterol crystals, ATP, or other danger signals — drives inflammasome assembly, caspase-1 activation, and the release of IL-1β that fuels the inflammatory arm of the fusion loop. Source: Kelley N, et al. "The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation." Int J Mol Sci. 2019;20(13):3328. PMC6651423. Used under CC BY 4.0 license.
Breaking the Loop: Three Nodes of Intervention
If the problem is a cycle, the solution must target multiple nodes. Here's how we currently think about breaking the loop:
| Node of Intervention | Target | Drug Class/Agent | Trial Evidence |
|---|---|---|---|
| Cholesterol Substrate | Reduce LDL delivery to vessel wall | Statins, ezetimibe, PCSK9i, bempedoic acid | 4S, PROVE IT, FOURIER |
| Crystal Formation | Prevent cholesterol crystallization at membrane level | Icosapent ethyl (EPA), high-dose omega-3 | REDUCE-IT, Mason membrane studies |
| Inflammatory Cascade | Dampen NLRP3 activation and IL-1β signaling | Colchicine, IL-1β inhibitors | COLCOT, LoDoCo2, CANTOS |
Dual-Action Agents on the Horizon
Several emerging therapies target multiple nodes simultaneously:
- Bempedoic acid: An ATP-citrate lyase (ACL) inhibitor that lowers LDL-C and reduces hs-CRP, suggesting dual anti-lipid and anti-inflammatory effects — a true dual-pathway agent.
- GLP-1 receptor agonists: Originally developed for glucose control, these agents have shown surprising cardioprotective and anti-inflammatory properties — and they help with weight loss, which itself reduces inflammation.
- NLRP3 inflammasome inhibitors: Currently in clinical development, these molecules directly target the molecular complex that links cholesterol crystals to IL-1β release.
The REDUCE-IT Question: The Right Drug for the Wrong Reason?
No discussion of the fusion hypothesis can ignore REDUCE-IT, perhaps the most controversial and enlightening trial in recent cardiovascular history.
Many experts believe — and we are in this camp — that the REDUCE-IT Trial may have merely identified a high-risk patient population, as its positive impact had no clear relation to triglyceride lowering. If that's true, it changes everything about how we interpret the trial's findings.
REDUCE-IT at a Glance
Design: 8,179 patients with prior MI or stable CAD, on statin therapy, with triglycerides 135–499 mg/dL and LDL-C 41–100 mg/dL.
Intervention: Icosapent ethyl 4g daily vs. mineral oil placebo.
Result: 25% relative risk reduction in cardiovascular death, MI, stroke, coronary revascularization, or unstable angina. Median follow-up 4.9 years.
The Puzzle: The benefit showed almost no correlation with triglyceride lowering — the trial's nominal target.
Here's what makes REDUCE-IT so important to the fusion hypothesis:
REDUCE-IT enrolled a specific population: patients with elevated triglycerides despite statin therapy. Why might these patients be special? One interpretation: patients with elevated triglycerides may represent a more active cholesterol-inflammation fusion loop. They may have higher oxidized and aggregated LDL (agLDL). They may form cholesterol crystals more readily. They may have a more primed NLRP3 inflammasome. In other words, their elevated triglycerides may be a marker of an unusually dangerous inflammatory-lipid interaction.
If that's true, then EPA's benefit might have less to do with lowering triglycerides and more to do with disrupting that interaction directly.
And this is where the Mason mechanism becomes crucial. EPA — the long-chain omega-3 fatty acid — can physically intercalate into cell membranes and alter the organization of cholesterol domains. It may prevent the cholesterol crystallization that triggers NLRP3 activation. In essence, EPA may work by disrupting the fusion cycle at the membrane level, not by lowering any number on a lab slip.
This interpretation is supported by the STRENGTH trial, which tested a combination of EPA and DHA (another omega-3 fat). Unlike REDUCE-IT, STRENGTH showed no benefit. Why? One possibility: DHA may actually increase the formation of lipid microdomains rich in cholesterol — potentially amplifying the very crystallization problem we're trying to prevent. EPA without DHA works. EPA plus DHA doesn't. The mechanism matters.
The JUPITER Connection: Sound Familiar?
REDUCE-IT isn't the only trial that enrolled patients who, on closer inspection, may have been identified by a fusion-prone phenotype rather than for a single biomarker target.
JUPITER (2008)
Design: 17,802 apparently healthy individuals with LDL-C <130 mg/dL but hs-CRP ≥2.0 mg/L, randomized to rosuvastatin 20 mg vs. placebo.
Result: 44% reduction in cardiovascular events. The trial was stopped early for overwhelming benefit. Among those who achieved both LDL-C <70 mg/dL and hs-CRP <2 mg/L, the reduction was 65% — a dual-target response that would later echo in the fusion hypothesis framework.
The Controversy: Critics argued that these patients weren't as "healthy" as they appeared. Many had features of the atherogenic triad — elevated triglycerides, low HDL, and increased small dense LDL particles — markers of insulin resistance and cardiometabolic disease. Their elevated CRP may have reflected underlying adiposopathy as much as vascular inflammation. In other words, JUPITER may have enrolled a population already caught in an early fusion loop — metabolically unhealthy individuals whose high CRP was a signal of both adipose-driven and vascular inflammation.
The JUPITER parallel is striking: like REDUCE-IT, it enrolled a population defined by a biomarker that may have been a surrogate for something deeper. Both trials achieved outsized benefits. Both enrolled patients with features suggestive of an active cholesterol-inflammation interaction. And both suggest that the real value of the biomarker — whether CRP or triglycerides — may be in identifying patients in whom the fusion loop is already running, not in targeting the biomarker itself.
What This Means for Your Patient — And Linda's Story
We may have to fundamentally shift how we think about secondary prevention after a heart attack.
For decades, the mantra has been simple: lower LDL cholesterol. And LDL lowering works — the trials prove it. But the fusion hypothesis suggests that in some patients, we may have to concede that there's a role for inflammation alone in driving recurrent events. Or at minimum, that both arms of the cycle need attention.
Here's how we're starting to think about it for patients like Linda:
The Fusion-Aware Risk Assessment
For Linda, this checklist led to a staged, thoughtful approach — not a single prescription change, but a sequential strategy that addressed different nodes of the cycle over time.
Step 1 — Targeting Inflammation: Given her recurrent event and hs-CRP of 2.5 mg/L, her cardiologist started colchicine 0.5 mg daily, supported by COLCOT and LoDoCo2 evidence. Three months later, her hs-CRP had drifted down to 1.5 mg/L. Was that the colchicine dampening NLRP3-driven inflammation? Was it continued weight loss reducing adiposopathy? Probably both. The point: the inflammatory signal was moving in the right direction.
Step 2 — Disrupting the Crystal Pathway: With triglycerides still at 195 mg/dL despite rosuvastatin 40 mg, her cardiologist added icosapent ethyl (Vascepa) 4g daily. The rationale wasn't just "lower the triglycerides" — it was the REDUCE-IT signal and the Mason membrane mechanism. If EPA can physically prevent cholesterol crystallization and NLRP3 activation at the membrane level, Linda's elevated triglycerides — likely a marker of a more active fusion loop — made her an ideal candidate.
Step 3 — The Plateau: Six months in, Linda's weight loss had stalled. She'd lost 12 pounds after her initial event and another 4 since the colchicine and dietary changes, but she'd hit a plateau. Her CRP was holding around 1.3 mg/L. Good — but was that as low as it could go? Her visceral adiposity, while improved, was still contributing inflammatory signals. Her A1c had crept back up to 6.4%.
Step 4 — Adding GLP-1 Receptor Agonist Therapy: Her cardiologist and endocrinologist jointly decided to initiate a GLP-1 receptor agonist — a class of drugs with demonstrated cardiovascular benefit, potent weight loss effects, and emerging evidence of anti-inflammatory properties. Over the next several months, Linda lost an additional 18 pounds. Her A1c dropped to 5.8%. And her hs-CRP fell below 0.5 mg/L.
Looking Ahead: The Next Chapter
The fusion hypothesis is still being tested. But several areas of active investigation suggest where the field is headed:
- NLRP3 inflammasome inhibitors: Several are in clinical development, with the first likely to reach the market within the next few years. These would represent the most direct pharmacologic attack on the cholesterol-crystallization-to-inflammation link.
- Lipoprotein(a)-lowering therapies: Lp(a), an LDL-like particle, is increasingly recognized as both a major cardiovascular risk factor and a potent driver of inflammation. New agents that lower Lp(a) may help address another source of fuel for the fusion loop.
- Imaging biomarkers: PET-CT imaging that can visualize inflammation (using FDG-PET) and atherosclerotic burden simultaneously may allow us to identify patients in whom the fusion loop is most active and target them for combination therapy.
- Precision risk stratification: The future may involve combining LDL-C levels, hs-CRP levels, imaging data, and genetic markers to create a more complete picture of an individual's risk — and then tailoring combination therapy accordingly.
The Bottom Line: A Hypothesis, Not an Imperative
It may be that heart disease doesn't play by one set of rules.
Cholesterol and inflammation may not be parallel problems — they may be co-conspirators locked in a self-reinforcing cycle. Breaking one arm of the loop slows the disease. Breaking both may stop it. The future of cardiovascular prevention may not be about choosing between lipid-lowering and anti-inflammatory therapy — it may be about knowing when and how to use both, for the right patient, at the right time.
And there remains the tantalizing possibility that driving LDL-C to extremely low levels may itself quiet the inflammatory arm — we just don't have the definitive data yet. We should remain humble about what we know and transparent about what we're still learning.
CardioAdvocate Rounds
Pull up a chair. We saved you a seat.
Real cases. Real evidence. What we're seeing, what we're missing, and why it matters — delivered to your inbox by a cardiologist who believes everyone deserves a seat at rounds.
Join Rounds →No spam. Unsubscribe anytime. We respect your inbox.
CardioAdvocate helps people understand what matters — and how to speak up about it.
Disclaimer: This article reflects current evidence and expert interpretation as of March 2026. The cholesterol-inflammation fusion hypothesis is an emerging framework based on mechanistic studies and clinical trials, but it is not yet consensus science. Treatment decisions should always be made in consultation with your healthcare provider based on individual risk factors, current medications, and personal values. The evidence cited represents the best available science at the time of writing and may evolve as new data emerges. CardioAdvocate does not endorse any specific medication or treatment without consideration of individual patient circumstances.