
Sticky Situation: Aortic Stenosis
Sticky Situation — Aortic Stenosis: When Prevention Meets Genetics
A forward-looking perspective on where medicine, genetics, and early prevention are heading
Case Presentation
A Question That Changed Everything
A 72-year-old retired engineer arrives in clinic with a 6-month history of progressive shortness of breath on exertion. He can no longer climb the stairs to his bedroom without stopping. His primary care physician heard a systolic murmur and referred him to the cardiologist.
An echocardiogram reveals the diagnosis: severe calcific aortic stenosis. The aortic valve area is just 0.8 cm² (normal is 3–4 cm²), and the mean pressure gradient is 48 mmHg. He is at class C2 for intervention, and the structural heart team discusses transcatheter aortic valve replacement (TAVR) as the definitive treatment.
But during his extended history, something emerges. His Lp(a)—lipoprotein(a)—was finally checked for the first time in his life. The result: 285 nmol/L (severely elevated; normal is <50). His LDL-C was 165 mg/dL in his 40s. He took a statin briefly, then his primary doctor discontinued it because "your cholesterol was fine." No family screening was ever performed—even though his father underwent aortic valve replacement at age 68.
He asks his cardiologist: "Could this have been prevented?"
Flying Under the Radar
Aortic stenosis is the most common valvular heart disease in the developed world. For decades, we called it "degenerative valve disease"—as though it were simply the inevitable wear and tear of aging, no different from gray hair or wrinkles.
But that narrative is crumbling. The truth is more unsettling: aortic stenosis is not fundamentally different from atherosclerosis. It is lipid-driven disease.
The paradigm shift: The same lipoproteins that infiltrate coronary arteries—specifically lipoprotein(a) [Lp(a)] and low-density lipoprotein cholesterol (LDL-C)—also infiltrate the aortic valve. There, they trigger inflammation, trigger a cascade of immune activation, and ultimately drive calcification and stenosis.
For years, this was merely a biologically plausible hypothesis. But in February 2026, two landmark genome-wide association studies (GWAS) published simultaneously in Nature Genetics provided definitive genetic evidence of causation. These studies identified 166 and 241 genetic loci, respectively, that predispose to aortic stenosis—and many of those loci overlap with the very same genes that drive LDL-C metabolism (PCSK9, LDLR, APOE) and lipoprotein(a) biology.
Here is the trap: By the time aortic stenosis becomes symptomatic—breathlessness, chest pain, syncope, or a murmur on exam—the disease is already far advanced. The aortic valve is already calcified, fibrotic, and stenotic. At that stage, the pathophysiology has long since transitioned from a lipid-driven inflammatory process to a purely mechanical/calcific one. There is no FDA-approved drug therapy for aortic stenosis. The only treatment is structural: TAVR or surgical aortic valve replacement.
The Critical Window: The lipid infiltration that initiates aortic valve disease likely occurs in the 4th and 5th decades of life—perhaps even earlier in those with genetic predisposition. By the time symptoms appear at age 70, 80, or beyond, it is 25–40 years too late for lipid-lowering therapy to reverse the calcification. Medical treatment must target the disease at its origin, not at its end stage.
CardioAdvocate Checklist
Have these conversations with your care team:
Questions to Ask Your Care Team
About Family Risk
"My father had aortic valve replacement at age 68. Does that mean I should be screened? What should be screened?"
"Should my siblings and children be tested for Lp(a) given what we now know about its role in aortic valve disease?"
About Your Own Risk
"Could my elevated Lp(a) be contributing to valve disease in the same way it increases my heart attack risk?"
"I've been told I have a valve problem but no symptoms yet. What does it mean if statins haven't helped in clinical trials? What IS being studied?"
About Prevention
"Is there anything I can do NOW—lifestyle changes, medications, supplements, diet—that could slow the progression of valve disease if I have early stenosis?"
"Should I be referred to a specialized trial or clinic focused on early lipid management for valve protection?"
Deep Dive: The Science Behind the Paradigm Shift
Why Statin Trials Failed — The "Too Little, Too Late" Problem
In the 2000s, cardiologists hypothesized that if cholesterol drives valve disease the same way it drives coronary disease, perhaps statins could halt or reverse aortic stenosis progression. Three major randomized controlled trials were launched:
SALTIRE (NEJM, 2005): 155 patients with mild-to-moderate aortic stenosis were randomized to high-dose atorvastatin (80 mg/day) versus placebo. After 2 years, there was no significant difference in the rate of aortic valve area decline. The trial failed.
SEAS (NEJM, 2008): 1,873 patients with mild-to-moderate asymptomatic aortic stenosis received simvastatin 40 mg plus ezetimibe 10 mg versus placebo. The primary composite endpoint (major cardiovascular events including aortic valve events and ischemic events) was not significantly reduced (HR 0.96, p=0.59). Notably, ischemic cardiovascular events were significantly reduced (HR 0.78, p=0.02) — but aortic valve events were completely unaffected (p=0.97). The valve disease marched on regardless of lipid lowering.
ASTRONOMER (Circulation, 2010): 269 patients with mild-to-moderate aortic stenosis were randomized to rosuvastatin versus placebo. There was no difference in the rate of aortic valve area decline. The trial failed.
Why did they all fail? The insight is crucial: These trials enrolled patients with established mild-to-moderate stenosis. The aortic valve in these patients already shows calcification, fibrosis, and early stenosis on imaging. By that point, the disease has already been underway for 20–30 years. The initial lipid-driven infiltration occurred decades earlier, when the patient was in their 40s or 50s, often without any screening or aggressive treatment. Treating late-stage calcific disease with statins is biologically naive—it is like trying to reverse a kidney stone with acid-lowering therapy.
Shared Pathophysiology: From Atherosclerosis to Aortic Valve Disease
For decades, aortic stenosis and coronary atherosclerosis were treated as fundamentally different diseases — one "degenerative," the other inflammatory. We now know they share a remarkably similar pathophysiological cascade:
Step 1: Lipid Infiltration. LDL particles and Lp(a) particles penetrate the endothelial surface of the aortic valve leaflets — just as they penetrate coronary artery walls. This is the initiating event, and it begins silently, often decades before symptoms.
Step 2: Oxidation and Immune Activation. Once trapped in the valve tissue, lipoproteins undergo oxidation. Oxidized phospholipids (OxPL) — particularly those carried on Lp(a) particles — trigger a potent inflammatory response. Macrophages and T-cells infiltrate the valve, releasing cytokines (IL-6, TNF-α) that perpetuate the inflammatory cycle.
Step 3: Osteogenic Transformation. Here is where the aortic valve diverges from coronary disease. The inflammatory milieu triggers valve interstitial cells (VICs) to undergo osteogenic differentiation — they literally transform into bone-forming cells. This is a one-way street: once VICs begin producing calcium, the process becomes self-sustaining.
Step 4: Calcification Begets Calcification. Calcium deposition stiffens the valve leaflets, altering the mechanical stress distribution. The increased stress accelerates further calcification in a vicious cycle. At this stage, the disease has transitioned from a lipid-driven, potentially treatable process to a calcification-driven, mechanically irreversible one. This is why statins failed in clinical trials — they were given too late, after the transition had already occurred.
📊 The Molecular Link: A landmark study by Thanassoulis et al. (NEJM, 2013) demonstrated that elevated Lp(a) is independently associated with aortic valve calcification in the general population. Subsequent work has shown that OxPL carried on Lp(a) directly stimulate osteogenic differentiation of valve interstitial cells — providing the mechanistic explanation for why Lp(a) causes valve disease, not just coronary disease.
LDL-C is also causal — not just Lp(a). Mendelian randomization analyses from the 2026 GWAS studies confirmed that both elevated Lp(a) and elevated LDL-C independently and causally increase the risk of aortic stenosis. This means aggressive LDL-C lowering matters for valve protection even in patients without elevated Lp(a) — the same cumulative LDL exposure concept that drives coronary atherosclerosis applies to the aortic valve.
🔍 The Critical Distinction: In coronary arteries, atherosclerotic plaques can be stabilized and even regressed with aggressive lipid lowering. In the aortic valve, once calcification passes a tipping point, the disease becomes self-propagating and irreversible without mechanical intervention. This is precisely why early lipid management is so critical for valve protection — you must intervene during the lipid-driven phase, before the calcification cascade becomes autonomous.
Bicuspid Aortic Valve: The Hidden Accelerator
A bicuspid aortic valve (BAV) is the most common congenital cardiac anomaly, affecting approximately 1–2% of the population — roughly 1 in 50 to 1 in 100 people. Instead of the normal three leaflets (tricuspid), the aortic valve has only two, often with a raphe (ridge) where the third leaflet would have formed.
Why does BAV matter for aortic stenosis? The answer lies in fluid dynamics. A bicuspid valve creates turbulent, non-laminar flow across its surface. This abnormal flow pattern generates increased mechanical shear stress on the valve leaflets — and shear stress is a powerful accelerator of the lipid infiltration–inflammation–calcification cascade described above.
The result: patients with BAV develop clinically significant aortic stenosis 10–20 years earlier than those with normal tricuspid valves. A tricuspid valve patient might develop severe AS in their 70s or 80s; a BAV patient may face it in their 50s or 60s — or even earlier if compounded by genetic lipid disorders.
📊 The Double Hit: BAV + elevated Lp(a) or elevated LDL-C represents a particularly dangerous combination. The valve is already under mechanical stress from turbulent flow, making it more vulnerable to lipid deposition. Add a genetic lipid disorder on top, and the calcification cascade accelerates dramatically. The 2020 ACC/AHA Valvular Heart Disease Guidelines recommend echocardiographic surveillance of BAV patients, but lipid management in this population remains underemphasized.
BAV is heritable. First-degree relatives of BAV patients have a 5–10× higher prevalence of BAV compared to the general population. Current guidelines recommend screening first-degree relatives with echocardiography. BAV is also associated with aortopathy — dilation of the ascending aorta — which carries its own risks (aneurysm, dissection) independent of valve stenosis.
The clinical implication is clear: if you have a bicuspid aortic valve, aggressive lipid management is not optional — it is essential. Your valve is already mechanically compromised; the last thing it needs is decades of lipid bombardment on top of the turbulent flow stress.
Coronary Artery Calcium Scans: Don't Ignore the Valve
Here is a clinical pearl that is routinely overlooked: every coronary artery calcium (CAC) CT scan captures the aortic valve. The aortic valve sits in the field of view of the CT scanner, and aortic valve calcification (AVC) is often clearly visible — yet it frequently goes unreported.
This is a missed opportunity of enormous proportions.
Aortic valve calcification on a CAC scan is a red flag — particularly in a younger patient (under age 60). In a young person, AVC should immediately raise suspicion for:
- Bicuspid aortic valve — which causes earlier calcification onset due to turbulent flow
- Elevated Lp(a) — a genetic driver of valve calcification via oxidized phospholipids
- Elevated LDL-C or familial hypercholesterolemia — cumulative lipid exposure driving valve disease
The same applies to any chest CT — not just dedicated CAC scans. Lung cancer screening low-dose CT (LDCT), pulmonary embolism protocol CT angiography, pre-operative chest CTs, and routine thoracic imaging all capture the aortic valve. When calcification is seen on the valve, it must be reported and should trigger a clinical workup.
What should happen when AVC is found incidentally:
- Echocardiography to assess valve morphology (bicuspid vs. tricuspid) and hemodynamic severity
- Lp(a) measurement — if never checked
- Comprehensive lipid panel review including ApoB
- Family history assessment for valve disease, premature ASCVD, and known lipid disorders
- Referral to a valvular heart disease or preventive cardiology specialist if appropriate
The tragedy is that incidental AVC findings are often buried in radiology reports or dismissed as "age-related degenerative changes" — even in patients in their 40s and 50s. As the field evolves, we expect AVC reporting on all chest CTs to become standard practice — turning every CT scan into a screening opportunity for early valve disease.
The GWAS Breakthrough: February 2026
In February 2026, two landmark studies transformed our understanding of aortic stenosis genetics:
Study 1: Kany et al., Nature Genetics (2026)
Kany et al., Nature Genetics 2026: This study used artificial intelligence and deep learning to analyze 59,571 MRI cardiac scans from UK Biobank participants. The researchers developed a machine-learning algorithm to quantify aortic root calcification and then performed a genome-wide association study (GWAS) on the imaging phenotype.
Result: 166 genetic loci were identified as associated with aortic valve calcification. Strikingly, many of these loci overlapped with pathways controlling LDL cholesterol, lipoprotein(a), and atherosclerotic cardiovascular disease. Key hits included:
- PCSK9 — A critical regulator of LDL receptor density and plasma LDL-C levels. PCSK9 inhibitors (proprotein convertase subtilisin/kexin type 9 inhibitors) are among the most potent LDL-lowering drugs available.
- LDLR — The LDL receptor itself, fundamental to cholesterol metabolism.
- APOE — Apolipoprotein E, a key determinant of lipoprotein metabolism.
- Multiple other loci involved in lipid transport, inflammation, and calcium homeostasis.
Study 2: Small et al., Nature Genetics (2026)
Small et al., Nature Genetics 2026: This multi-ancestry GWAS study was the largest ever conducted on aortic stenosis risk. The researchers analyzed 86,864 aortic stenosis cases among 2.85 million individuals from multiple ancestry groups and performed transcriptome analysis of human aortic valve tissue.
Result: 241 autosomal genetic risk loci + 3 X chromosome loci were identified. Of these, 54 were newly discovered genes not previously implicated in aortic stenosis. Pathway analysis revealed enrichment for:
- Lipid metabolism and lipoprotein biology
- Inflammatory pathways (IL-6, TNF-α signaling)
- Calcium homeostasis and bone mineralization
- Endothelial function and vascular biology
Most compellingly, Mendelian randomization analysis provided causal evidence that both elevated Lp(a) and elevated LDL-C increase the risk of aortic stenosis. This is not mere association; it is causation.
Commentary in Nature Cardiovascular Research: "These findings provide a strong foundation for the future development of medical therapy targeting lipid pathways in aortic stenosis. The convergence of genetic, imaging, and causal evidence suggests that early aggressive lipid management—particularly in genetically susceptible individuals—may prevent the initiation of valve disease altogether."
The Case for Primordial Prevention
If Lp(a) and LDL-C truly cause aortic stenosis—just as they cause atherosclerotic plaques in coronary arteries—then the logical corollary follows: Early, aggressive lipid management could theoretically prevent aortic stenosis from ever developing in the first place.
This is the concept of primordial prevention—preventing the disease before it starts, by eliminating the causal risk factor early in life. In coronary artery disease, primordial prevention means aggressive LDL-C and Lp(a) lowering starting in the 20s or 30s for high-risk individuals, long before atherosclerosis becomes symptomatic.
The same principle should apply to aortic stenosis:
- Screen for Lp(a) early. A single Lp(a) measurement at age 20 or 30 identifies lifelong risk. High Lp(a) is heritable and does not change with lifestyle modifications. Genetic predisposition is permanent.
- Maintain aggressive LDL-C control throughout adulthood. The concept of "lifetime cumulative LDL-C exposure" is as valid for the aortic valve as it is for coronary arteries. Decades of mild-to-moderate LDL-C elevation (130–160 mg/dL) likely sets the stage for valve disease just as it does for heart attacks.
- Consider emerging Lp(a)-lowering therapies. Multiple drug candidates are in clinical trials (periprocedural antisense oligonucleotide therapy, small molecules, gene therapy). If these therapies prove effective in preventing heart attacks, the next natural step would be to test them for valve disease prevention.
- Identify genetically high-risk families. Polygenic risk scores for aortic stenosis may soon be available. Cascade screening of relatives—just as is done for familial hypercholesterolemia—could identify those who need most aggressive lipid management.
Understanding Your Echocardiogram: How We Monitor Aortic Stenosis
The echocardiogram is the cornerstone of aortic stenosis surveillance. It's a painless ultrasound that shows us the valve in real time — how it opens, how it closes, how thick and calcified the leaflets are, and how the heart muscle is responding to the obstruction. Here's what your cardiologist is looking at:
Peak Velocity and Mean Gradient. As the valve narrows, blood accelerates through the smaller opening — like water through a pinched garden hose. Peak velocity above 4 m/s and mean gradient above 40 mmHg indicate severe stenosis. Your cardiologist tracks these numbers over time; a rapid rate of progression (increase of >0.3 m/s per year) is a red flag that intervention may be needed sooner rather than later.
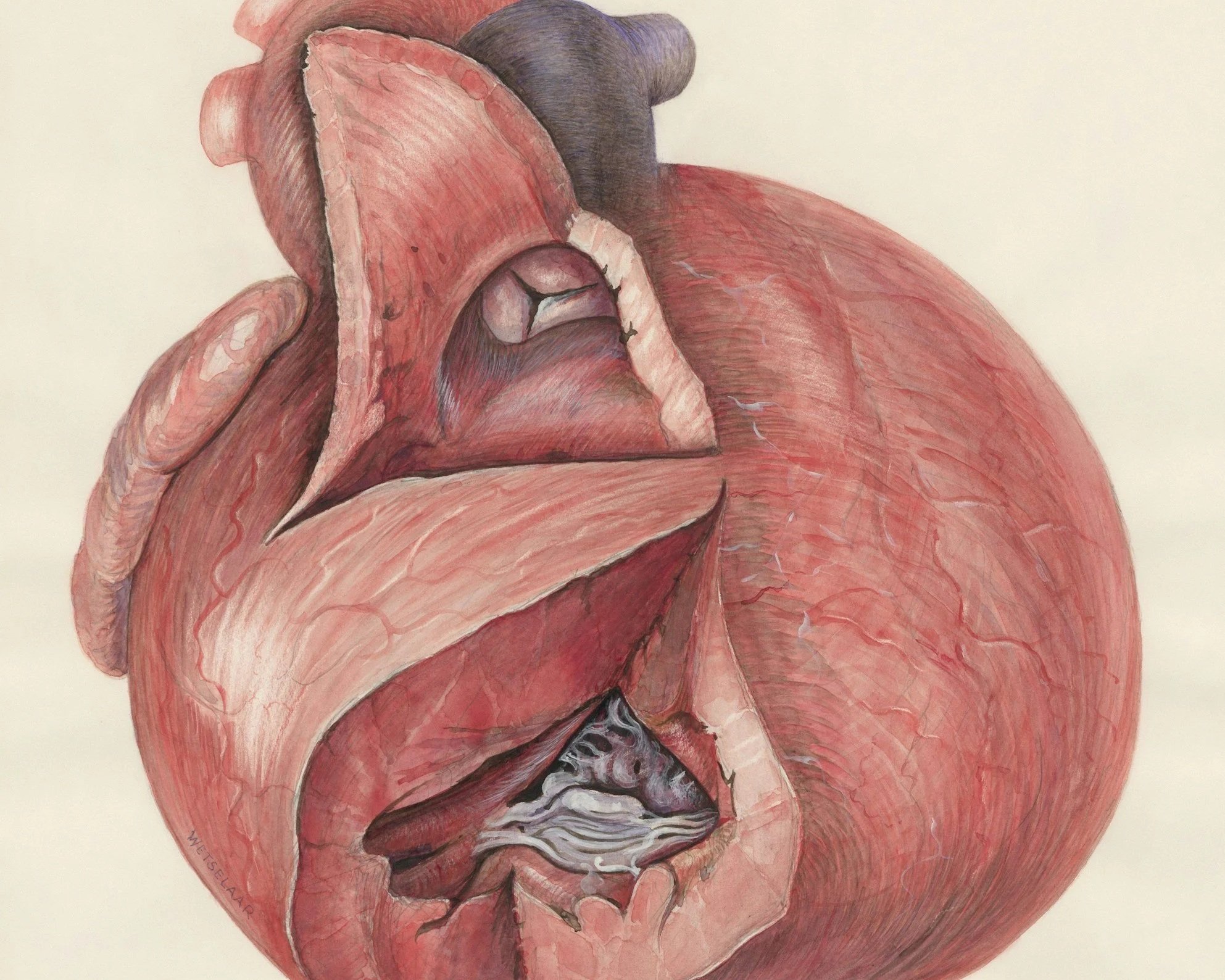
Aortic Valve Area (AVA). A normal aortic valve opens to 3-4 cm². Severe aortic stenosis is defined as an AVA less than 1.0 cm² — meaning 75% of the valve's opening has been lost to disease. Below 0.6 cm², we call it "critical" stenosis.
Left Ventricular Response. The heart doesn't take this obstruction lying down. Initially, the left ventricle thickens (hypertrophy) to generate more force. Your echo shows wall thickness, chamber size, ejection fraction, and diastolic function — all of which tell the story of how well the heart is compensating. When compensation fails — the ventricle dilates, EF drops, or filling pressures rise — the clock is ticking toward intervention.
Serial Monitoring Schedule. The 2020 ACC/AHA Valvular Heart Disease Guidelines recommend serial echocardiography based on severity: every 3-5 years for mild AS (peak velocity 2.0-2.9 m/s), every 1-2 years for moderate AS (3.0-3.9 m/s), and every 6-12 months for severe AS (≥4.0 m/s). But these intervals should be shortened when the rate of progression is rapid or when symptoms are ambiguous.
When the Valve Needs Replacing: SAVR vs. TAVR
For decades, aortic stenosis was the textbook example of a "mechanical problem requiring a mechanical solution." When the valve became critically narrow, the only option was surgical aortic valve replacement (SAVR) — open-heart surgery, sternotomy, cardiopulmonary bypass, weeks of recovery. SAVR remains a remarkably effective operation with excellent long-term durability, particularly with modern bioprosthetic valves. For younger patients (under 65-70), those with bicuspid aortic valves, concurrent coronary artery disease requiring bypass, or aortic root dilation, SAVR is often still the preferred approach.
Then came TAVR. First approved in the US in 2011 for inoperable patients, TAVR has undergone one of the most dramatic expansions in the history of cardiovascular medicine. Within a decade, it moved from a last-resort option for the sickest patients to a first-line consideration across the entire risk spectrum.
The Landmark Trials. The PARTNER trials (using Edwards SAPIEN valves) and Evolut trials (using Medtronic CoreValve) systematically demonstrated TAVR's safety and efficacy across risk categories. The PARTNER 3 trial (NEJM, 2019) showed TAVR was superior to SAVR in low-risk patients at 1 year. The Evolut Low Risk trial (NEJM, 2019) demonstrated non-inferiority. Now, with 7-year follow-up data from PARTNER 3 (TCT 2025) and 5-year data from Evolut Low Risk (ACC 2025), the durability story is reassuring: mortality, stroke, and valve function are comparable between TAVR and SAVR out to 5-7 years.
The Durability Question. The honest nuance: TAVR valves are still younger technology than surgical bioprostheses. Structural valve deterioration (SVD) — the gradual failure of bioprosthetic leaflets — remains a concern, particularly for younger patients who will stress these valves for decades. The NOTION-2 trial 3-year results (Circulation, 2025) in younger low-risk patients (ages 60-75) showed a numerically higher composite endpoint with TAVR vs. SAVR (16.1% vs. 12.6%), though this was not statistically significant in a relatively small, exploratory cohort. TAVR also carries higher rates of pacemaker implantation and paravalvular leak compared to SAVR. The reality is that we need 10-15 year data before we can definitively say TAVR is equivalent to SAVR in durability for patients in their 50s and 60s.
The EARLY TAVR Revolution: Intervening Before Catastrophe
For generations, the textbook teaching was clear: don't replace the valve until symptoms appear. Wait for the chest pain, the syncope, the breathlessness climbing stairs. This watchful waiting strategy made sense when valve replacement meant cracking open the chest — the risk of the surgery had to be justified by the severity of symptoms.
But TAVR has changed the risk calculus. With procedural mortality now under 1% in experienced centers and most patients going home the next day, the question is no longer "can we intervene safely?" but "are we waiting too long?"
The EARLY TAVR trial (NEJM, 2024) answered this definitively. In 901 patients with asymptomatic severe aortic stenosis randomized to early TAVR vs. clinical surveillance:
- The primary endpoint (death, stroke, or unplanned cardiovascular hospitalization) occurred in 26.8% of early TAVR patients vs. 45.3% in surveillance (HR 0.50, p<0.001) — a 50% relative risk reduction.
- Unplanned cardiovascular hospitalizations were cut in half: 20.9% vs. 41.7%.
- Mortality was similar (8.4% vs. 9.2%), but follow-up was only 3.8 years — the mortality curves are expected to separate further.
- Early TAVR prevented decline in quality of life and preserved left ventricular function.
The Horizon: From Mechanical Fix to Medical Prevention
Here's where the story gets exciting — and, as Dr. Riddock would say, a bit speculative. For over a century, aortic stenosis has been framed as a "mechanical problem requiring a mechanical solution." The valve calcifies, it narrows, you replace it. Full stop. There was no medical therapy. Statin trials in established AS (SEAS, SALTIRE, ASTRONOMER) all failed to slow progression.
But here's the critical insight those trials missed: they treated too late. By the time you have moderate-to-severe calcific AS, the disease has transitioned from an active lipid-inflammatory process to a self-perpetuating calcification cascade. Statins can't reverse calcium deposits any more than they can un-calcify a coronary artery. The hypothesis has always persisted that intervention during aortic sclerosis — the earliest stage, before significant calcification — might be a different story entirely.
Now, with Mendelian randomization proving that both LDL-C and Lp(a) are causally linked to aortic valve disease, we finally have the biological rationale to test this hypothesis properly. The emerging paradigm looks like this:
The Window of Opportunity: Aortic Sclerosis. When an echocardiogram first shows thickening of the aortic valve without obstruction — that's aortic sclerosis. It's present in ~25% of adults over 65. Most clinicians document it and move on. But if aortic valve disease truly shares the same lipid-driven, inflammatory pathophysiology as atherosclerosis (and the genetic evidence now strongly suggests it does), then aortic sclerosis is the "fatty streak" of the valve — and the ONLY window where aggressive lipid lowering might actually halt progression before calcification becomes self-perpetuating.
Lp(a)-Lowering Therapies: The Game Changer? The failed statin trials never addressed Lp(a) — they couldn't. Statins don't lower Lp(a). But emerging therapies like pelacarsen (an antisense oligonucleotide targeting apo(a) mRNA) and olpasiran (a small interfering RNA) can lower Lp(a) by 80-98%. The HORIZON trial is testing pelacarsen for cardiovascular outcomes. If these agents reduce valve disease progression in patients with elevated Lp(a) and early aortic sclerosis, we will have fundamentally changed the natural history of the disease.
The Speculative Leap — and Why It's Rational. Imagine a 45-year-old with elevated Lp(a) (say, 150 nmol/L) whose routine echocardiogram shows early aortic sclerosis. Today, we document it and recheck in a year. Tomorrow? We might start Lp(a)-lowering therapy, aggressively manage LDL-C to below 55 mg/dL, and monitor the valve with serial imaging — not to prepare for eventual valve replacement, but to prevent it from ever being needed. That's the paradigm shift: from treating end-stage valve disease with hardware to preventing it with pharmacology.
Where Medicine, Genetics, and Prevention Are Heading
The 2026 GWAS breakthroughs have opened a new frontier. Over the next 5–10 years, expect:
1. Drug Development Targeted at Genetic Pathways
If PCSK9, LDLR, and Lp(a) pathways are truly causal in aortic stenosis, pharmaceutical companies will develop next-generation therapies targeting these pathways specifically for valve disease. PCSK9 inhibitors and other emerging agents (inclisiran, periprocedural therapies, small molecule inhibitors) may soon have aortic stenosis prevention as an indication.
2. Polygenic Risk Scores for Aortic Stenosis
Using the 241 genetic loci identified in the February 2026 GWAS, researchers will develop polygenic risk scores to stratify individuals by genetic susceptibility to valve disease. A 30-year-old with a high polygenic risk score + elevated Lp(a) would be a candidate for aggressive, early lipid-lowering therapy.
3. Clinical Trials of Early Lipid Lowering
Ongoing trials like NCT05646381 (an Lp(a)-lowering trial) may eventually extend to studying whether lowering Lp(a) in the 4th and 5th decades prevents aortic stenosis from ever developing. Such trials would take 10–15 years but could fundamentally shift prevention paradigms.
4. Precision Medicine Approach
The future patient with elevated Lp(a) or familial hypercholesterolemia will be counseled not only about coronary heart disease risk but also about aortic stenosis risk. Screening echocardiography may become standard in high-risk genetic cohorts starting in the 5th decade to catch early, asymptomatic disease.
5. Integration of Genetics into Primary Care
As the field evolves, genetic screening for aortic stenosis predisposition may become part of routine cardiovascular risk assessment—similar to how familial hypercholesterolemia cascading genetic screening is now recommended by guidelines.
The Unsettling Truth: For patients like the retired engineer in our case presentation, the disease could potentially have been prevented—or at minimum, its progression slowed to the point where intervention is unnecessary in a person's lifetime. That he was never screened for Lp(a), never aggressively treated for LDL-C, and never underwent echocardiography despite a positive family history is a systems failure. As genetics and prevention converge, such oversights should become rarer.
Bottom Line
1. Aortic stenosis is not simply "wear and tear." It is a lipid-driven, inflammatory disease driven by the same causal factors—Lp(a) and LDL-C—that cause heart attacks and strokes.
2. The 2026 GWAS studies provide definitive genetic proof of causation. We now have 241 loci and Mendelian randomization evidence. The old "degenerative valve disease" narrative is gone.
3. Failed statin trials teach us timing is everything. Treating established calcific aortic stenosis is too late. The disease initiated decades earlier, during a person's 40s and 50s, when lipid infiltration occurred silently in the valve.
4. The case for primordial prevention is compelling. Early, aggressive LDL-C control + Lp(a) awareness starting in the 20s and 30s for high-risk individuals could potentially prevent aortic stenosis from ever developing.
5. Screening matters. A single Lp(a) measurement at age 20–30 identifies lifelong risk. Echocardiography screening in those with elevated Lp(a), familial hypercholesterolemia, or a family history of valve disease should become standard practice.
6. Family history is a red flag. If a parent, sibling, or child has had aortic valve disease, the others in the family should be screened for both Lp(a) and early valve disease.
7. The frontier of preventive cardiology is here. We can no longer accept aortic stenosis as inevitable. Treat the disease early, treat it aggressively, and treat it long—before the first symptom ever appears.
8. TAVR has changed the game. What was once only treatable with open-heart surgery can now be addressed with a catheter-based procedure going home the next day. Long-term data at 5-7 years shows durable results — but the decision between TAVR and SAVR must be individualized by a Heart Team.
9. Don't wait for symptoms to act. The EARLY TAVR trial proved that early intervention in asymptomatic severe AS reduces cardiovascular events by 50%. The old "watch and wait" approach was built for a different era.
10. The biggest paradigm shift may be medical, not mechanical. With Mendelian randomization proving LDL-C and Lp(a) are causal, and Lp(a)-lowering agents on the horizon, the future of aortic stenosis may be prevention with pharmacology — not replacement with hardware.
Related CardioAdvocate Phenotypes
Explore these interconnected topics for a deeper understanding of how lipids, genetics, and prevention converge:
Little Napoleon Complex — Lipoprotein(a) Understand why Lp(a) is the "forgotten" risk factor that drives both atherosclerosis and valve disease.
Follow the Leader — Lipid Guidelines How lipid management strategies are evolving to address not just heart attacks, but valve disease prevention.
There's an App for That — Risk Calculators Emerging polygenic risk scores and genetic algorithms for predicting aortic stenosis susceptibility.
A Picture Is Worth a Thousand Words — Coronary Artery Calcium Why calcification in the coronary arteries often signals calcification in the aortic valve—and what to do about it.
Hiding in Plain Sight — Familial Hypercholesterolemia When genetic predisposition to high cholesterol increases the risk not only of early MI, but also of early aortic stenosis.